Method Development
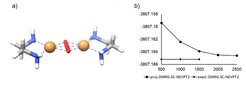
In the last decade the ab initio density matrix renormalization group (DMRG) has been shown to provide a reasonable and accurate alternative to complete active space (CAS) methods as basis for molecular multireference calculations. It can be regarded as an approximation to the exact diagonalization of the large Hamiltonian matrix in the basis of many-electron wavefunctions within the active orbital space. A great advantage of DMRG is that it approximately solves a problem whose complexity scales exponentially with increasing system size by optimizing only a polynomial number of parameters. Owing to this favorable behavior DMRG is able to treat large active spaces on the order of 20-80 orbitals. However, quantitative accuracy is only reached if dynamic electron correlation effects are considered, too. Therefore we have developed a novel approach to the combination of DMRG and strongly contracted second order N-electron valence perturbation theory (SC-NEVPT2) for quantum chemical multireference calculations. The main objective of this approach is to lower the cost to treat systems with large active spaces and large orbital spaces with a moderate and controllable accuracy. As demonstrated for a dimeric Cu cluster the total energy (DMRG + SC-NEVPT2) converges rapidly and smoothly towards the exact value with increasing bond dimension which is a user-defined accuracy parameter. However, the existing method considerably suffers from the comparably high bond dimensions required to achieve chemical accuracy. In this regard, the most promising route forward for the future may be to sacrifice some of the constraints used in the projection approximation associated with the formally low scaling.

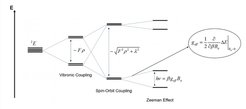
Furthermore we have developed an approach to describe spin-orbit coupling (SOC) on top of a regular Born-Oppenheimer DMRG calculation in the framework of quasi-degenerate perturbation theory (QDPT). This approach accounts for SOC effects on the many-electron level and can thus be thought of as the molecular equivalent of atomic Russell-Saunders or LS coupling. With the spin-orbit coupled wavefunctions at hand the molecular g-tensors can be calculated in a rigororous and phenomenological way as proposed by Gerloch and McMeeking in 1975. Importantly, since the SOC matrix is fully diagonalized within a finite set of many-electron states, our approach is able to produce qualitatively and quantitatively correct results even for systems with a near-degenerate ground state. For example, the behavior of the molecular g-values of a Mo(III) trisamidoamine catalyst as it is distorted along its Jahn-Teller axis is correctly reproduced. In contrast, regular linear-response type or single reference perturbation theory methods are bound to fail in these cases. Our method will serve to investigate the magnetic properties of complex molecular systems that cannot be properly treated by conventional DFT or CASSCF based approaches. Moreover, future developments might concern the inclusion of SOC to excited states outside the active space regime and the combination with schemes to incorporate dynamic electron correlation.