Applications
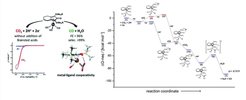
Among the numerous methods for the activation of carbon dioxide and its use as C1-building block, its electrocatalytic reduction constitutes a promising approach. In particular, the electrogeneration of carbon monoxide is attractive, since CO is used as a key feedstock in industry for a wide range of organic transformations. Due to the high overpotential and a variety of possible reaction pathways, a selective electroreduction of CO2 to CO is not trivial. A general possibility to overcome the issue of kinetic inhibition of an electrochemical reaction is the use of homogeneous redox-active catalysts in an indirect electrolysis. Under given conditions (substrate concentration, electrode material, electrolyte, etc.), the employment of such redox catalysts can lower kinetic barriers and often leads to an increased and/or entirely different selectivity. Recently, the groups of Dr. Robert Francke and Prof. Dr. Matthias Beller identified a cyclopentadienone iron(0) complexes as molecular catalysts for the electroreduction of CO2. It effectively converts CO2 into CO with two electrons and two protons to yield H2O. Interestingly, the reaction occurs without the addition of any acid. With the help of our quantum chemical computations it was possible to propose a reasonable reaction mechanism that it is in good agreement with the experimental results. According to our results, binding of CO2 to the catalyst in the desired η1 fashion via its C-atom can only be observed after the catalyst has been twofold reduced. Furthermore, protonation of the catalyst prior to CO2 binding facilitates the binding process through formation of a hydrogen bond. Although the highest calculated barrier is associated to the breaking of the CO bond and the simultaneous formation of an OH bond, recent experimental findings indicate that the effectiveness of the reaction is controlled by a dimerization equilibrium of a crucial intermediate. Further experimental and computational studies on this system are currently conducted in the labs of Dr. Francke, Prof. Dr. Beller and Prof. Dr. Ludwig at the University of Rostock and the Leibniz Institute for Catalysis as well as our group.

Another recent computational study of our group involves the magnetic properties of two derivates of the molybdenum trisamidoamine (TAA) complex [Mo] (=(3,5-(2,4,6-i-Pr3C6H2)2C6H3NCH2CH2N)Mo). In particular, we investigated [Mo]NH and {[Mo]N}- by means of EPR spectroscopy as well as multireference quantum chemistry. Because of the degeneracy of the electronic ground states of both {[Mo]N}− and [Mo]NH, only multireference-based methods such as the complete active-space self-consistent field (CASSCF) and related methods provide a qualitatively correct description of the electronic ground state and vibronic coupling. The molecular g values of {[Mo]N}− and [Mo]NH exhibit large deviations from the free-electron value ge. Their actual values reflect the nature of the ground state as well the relative strengths of vibronic and spin–orbit coupling. In the course of the computational treatment, the utility and limitations of a formal two-state model that describes this competition between couplings are illustrated, and the implications of our results for the chemical reactivity of these states are discussed.