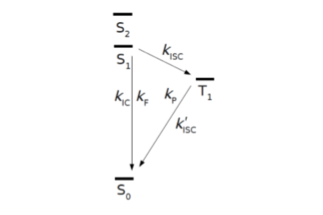
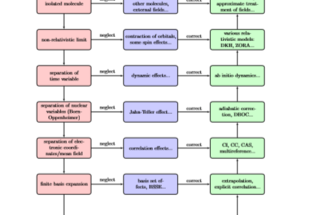
Theoretische Methodenentwicklung

Dr. Róbert Izsák hat im Jahr 2020 ans Middlebury College gewechselt. Nähere Informationen zu seiner jetzigen Tätigkeit finden Sie hier. Diese Seite dokumentiert die Forschung seiner Gruppe in der Abteilung Molekulare Theorie und Spektroskopie bis 2020.
What follows is a more general view on (wave function based) method development. As with any attempt that tries to give a bird’s eye view of a large research area, not everything can be fitted neatly into the scheme presented here. Nevertheless, the purpose of theoretical science is to build theories with quantitative predictive power from only a few principles taken for granted. These predictions can then be checked experimentally, the underlying assumption being that an ideally perfect experiment should match the predictions coming from an ideally ‘exact’ theory. In practice there are many obstacles both on the experimental and on the theoretical side. As there are no experiments without error, there are no theories without some flaws. A realistic goal is to keep these under control. In quantum chemistry, the ‘exact’ side of the coin basically consist of the principal assumptions of quantum theory and (special) relativity. The resulting equations are often not amenable to practical computations, and, therefore, a system of approximations is designed to arrive at something ‘computable’, something that can be practically evaluated. This role is taken by Hartree-Fock theory in quantum chemistry. In order to arrive at this theory, various approximations were made, but since in principle we know what we neglected, we know what effects this may cause, see Fig. 1. For example, if we ignore relativity, we cannot expect our method to account for the color of gold, which we then consider as a ‘relativistic effect’. On the other hand in principle we are also able to correct for what we neglected, i.e., we have control over the error we allow. This may not be an easy task, however. While Hartree-Fock theory recovers 98% of the total electronic energy, the recovery of the remaining few percents can be an extremely costly computational assignment, and yet a necessary one since most chemistry is affected by this ‘correlation energy’. For this reason, the purpose of method development is to devise efficient methods which allow us to accurately describe chemically interesting systems.